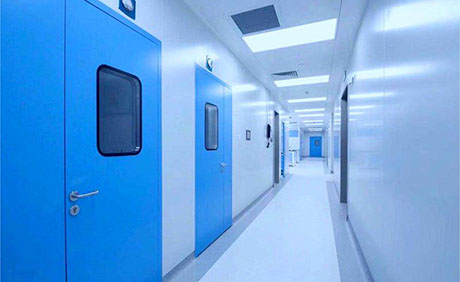
生物制药领域
净化工程解决方案
20余年净化工程建设总包经验 生物制药净化工程标杆企业
联系电话:186 6511 0000
懂生产、精工艺才是业主方所需的承建商,合景净化工程公司EPC一体化GMP工程总包服务,专家组全过程服务。合景服务GMP生物制药,中药制药(片剂、颗粒剂、丸剂、口服液、贴膏剂、中药饮片、原料药);化学制药(滴眼剂、水针剂、粉针剂、无菌原料药);生物制药(疫苗、抗生素、抗毒血清、动物提取、细胞工程);医药实验室(微生物室、理化实验室)等细分领域大型和超大型GMP生产车间的咨询、规划、设计、采购到建造EPC总承包,依托合景公司丰富的设计施工经验,打造优质GMP工程项目。










BRAND
DESIGN







QUALITY


洁净度测试


FFU测试


静压测试


照明度测试


温湿度测试


微生物测试
|
测试仪器:洁净度测试使用微粒计数器,仪器须经校正合格且仍在有效期限内及均须检附合格之校正档。
|
洁净度等级定级的粒径范围0.1~5.0μm。
|
|
测试步骤:确定采样点数量、绘制采样点布置图、确定测试仪器和采样量、现场采样、数据计算分析。
|
每个采样点最少采样时间为1分钟,且采样量应按照采用设备为准取值。
|
|
信赖度上限:任一无尘室或独立的隔间,若是取样点数小于10(2到9),每一取样点的平均值代表该点的微粒量测值。
|




|
检查空气管和探头有没堵塞
|
将探头的支撑脚紧贴于FFU表面、稳定。
|
|
由于是压差式风速计,因此读数时注意空气管不可被挤压,不可晃动。
|
每片FFU测量两点,计算平均值。
|
|
要求FFU风速上下相差不可大于20%。
|
|
静压差的测定应在洁净室(区)的风速、风量和送风均匀性检测合格后进行,并应在所有的门关闭时检测。
|
仪器宜采用各种微差压力计,仪表灵敏度应小于1.0Pa。
|
|
级别不同的洁净室区域与非洁净室区域之间的静压差,不小于5Pa。
|
洁净室与室外的静压差,不应小于10Pa。
|
|
打开空气洁净度等级高于百级的单向流洁净室门时,门内0.6米的室内工作区含尘浓度不应大于相应级别的含尘浓度限值。
|
|
照度的测定要求室内照度的测定必须在室温稳定,光源光输出稳定后再进行测定。
|
照度的测定方法:第一,照度的测点应距离地面0.85m, 按间距1~2m布点,测点距离墙1m。第二,记录实测值并计算总的平均照度。第三,照度测定一般是不含局部照明以外的一般照明。
|
|
合格标准: 应符合设计要求,根据(洁净厂房设计规范)的要求,洁净室内一般照明的均匀度不应小于0.7。
|
|
净化车间内的空气环境温度和相对湿度在检测之前,净化空调系统必须持续运作起码12个小时以上。
|
依据环境温度和相对湿度波动范围,检测宜持续开展8-48小时,每一次检测间隔时间不可超出30分钟。
|
|
净化车间内的测点通常应布置在距外墙表面层超出0.5米、距地0.8米的相同高度上;也可依据区域大小,各自布置在距地不一样高度的多个平面上。
|
|
通过自然沉降原理收集在空气中的生物粒子于培养基平皿中,让其繁殖至可见菌落进行计数,以菌落数判定环境活微生物数,以此评定洁净室(区)洁净度。
|
在洁净区墙面、工作台等的沉降菌测试采用适合规格条件的大豆酪蛋白琼脂培养基(TSA)配置的培养皿采样,在30℃~35℃培养箱中培养,时间不少于2d;采用沙氏培养基(SDA)配置的培养皿采样,在20℃~25℃培养箱中培养,时间不少于5d。
|
|
洁净室(区)的温度和相对湿度应与其生产及工艺要求相适应,温度在18℃~26℃,相对湿度在45%~65%为宜。
|
用肉眼对培养皿上的菌落直接计数、标记或者在菌落计数器上点计,然后用5~10倍放大镜检查,是否遗漏。
|
麦总:186 6511 0000
施工工地符合ISO14001和OHSAS18001体系要求,不会有任何对环境造成损害,影响周边可持续发展的事故发生
材料品管
工厂品管
运输品管
现场保管
材料检验
施工品管
设备检验
仪器校正
作业程序
静态管理
动态管理
安全管理
环境卫生
警勤管理
教育倡导
人员管理